Selecting and Comparing Genes with GSForge¶
Our goal in examining RNA-seq data sets often reduces to “feature selection” – to borrow a term from machine learning. Examining the count data should give us an idea of which genes correlate (by a given measure) with a phenotype of interest. We can then examine that ‘selected’ set more closely and form biological or chemical hypothesis that explain the expression-phenotype link. Unfortunately there is not a gold standard measure to preform this selection, and practicing researchers must juggle a number of different methods and results.
Enter GSForge:
A tool that helps collate and compare gene ‘selection’ results from a given method. Here we present a brief analysis
of a Oryza sativa cultivar set[1], with the explicit purpose of demonstrating GSForge.
from os import environ
from pathlib import Path
import numpy as np
import pandas as pd
import xarray as xr
import patsy
import GSForge as gsf
from sklearn.ensemble import RandomForestClassifier
from sklearn.preprocessing import quantile_transform
from sklearn import model_selection
from sklearn import linear_model
import umap
import umap.plot
import matplotlib.pyplot as plt
import seaborn as sns
import holoviews as hv
hv.extension('matplotlib')
# Declare paths.
OSF_PATH = Path(environ.get("GSFORGE_DEMO_DATA", default="~/GSForge_demo_data/")).expanduser().joinpath("osfstorage", "oryza_sativa")
RAW_COUNT_PATH = OSF_PATH.joinpath("GEMmakerGEMs", "rice_heat_drought.GEM.raw.txt")
HYDRO_LABEL_PATH = OSF_PATH.joinpath("GEMmakerGEMs", "raw_annotation_data", "PRJNA301554.hydroponic.annotations.txt")
SI_FILE_1_PATH = OSF_PATH.joinpath('GEMmakerGEMs', 'raw_annotation_data', 'TPC2016-00158-LSBR2_Supplemental_File_1.csv')
# Output paths.
TOUR_DGE = OSF_PATH.joinpath("GeneSetCollections", "tour_DGE")
TOUR_BORUTA = OSF_PATH.joinpath("GeneSetCollections", "tour_boruta")
LIT_DGE_GSC = OSF_PATH.joinpath("GeneSetCollections", "literature", "DGE")
LIT_TF = OSF_PATH.joinpath("GeneSetCollections", "literature", "TF")
/home/travis/virtualenv/python3.7.1/lib/python3.7/site-packages/holoviews/plotting/mpl/__init__.py:220: UserWarning: Trying to register the cmap 'fire' which already exists.
register_cmap("fire", cmap=fire_cmap)


import rpy2.rinterface_lib.callbacks
import logging
from rpy2.robjects import pandas2ri
%load_ext rpy2.ipython
pandas2ri.activate()
rpy2.rinterface_lib.callbacks.logger.setLevel(logging.ERROR) # Supresses verbose R output.
---------------------------------------------------------------------------
ModuleNotFoundError Traceback (most recent call last)
/tmp/ipykernel_5983/527480914.py in <module>
----> 1 import rpy2.rinterface_lib.callbacks
2 import logging
3 from rpy2.robjects import pandas2ri
4 get_ipython().run_line_magic('load_ext', 'rpy2.ipython')
5 pandas2ri.activate()
ModuleNotFoundError: No module named 'rpy2'
%%R
library("edgeR")
1. Create an Annotated Gene Expression Matrix¶
The AnnotatedGEM contains our expression matrix and any annotations that can be sample or gene indexed.
This object can be created directly from GEMmaker output and (properly formatted) annotation matrices.
See the notebook regarding AnnotatedGEM creation for more details.
Note
We used our groups GEMmaker workflow for the alignment and
quantification data presented here.
agem = gsf.AnnotatedGEM.from_files(
count_path=RAW_COUNT_PATH,
label_path=HYDRO_LABEL_PATH,
# These are the default arguments passed to from_files,
# to the individual calls to `pandas.read_csv`.
count_kwargs=dict(index_col=0, sep="\t"),
label_kwargs=dict(index_col=1, sep="\t"),
)
agem.data['time'] = agem.data['time'].to_series().str.split(' ', expand=True).iloc[:, 0].astype(int)
agem
<GSForge.AnnotatedGEM>
Name: AnnotatedGEM01619
Selected GEM Variable: 'counts'
Gene 55986
Sample 475
Under the hood this is a light-weight wrapper for the xarray.DataSet object, which does the heavy lifting.
The xarray.DataSet object is available under the .data attribute.
agem.data
<xarray.Dataset>
Dimensions: (Sample: 475, Gene: 55986)
Coordinates:
* Sample (Sample) object 'SRX1423934' ... 'SRX1424408'
* Gene (Gene) object 'LOC_Os06g05820' ... 'LOC_Os07g03418'
Data variables: (12/29)
BioSample (Sample) object 'SAMN04251848' ... 'SAMN04251607'
LoadDate (Sample) object '2015-11-20' ... '2015-11-19'
MBases (Sample) int64 4016 5202 4053 1166 ... 3098 3529 2922
MBytes (Sample) int64 2738 3652 2719 764 ... 1983 2370 1862
Run (Sample) object 'SRR2931040' ... 'SRR2931514'
SRA_Sample (Sample) object 'SRS1156722' ... 'SRS1156251'
... ...
Platform (Sample) object 'ILLUMINA' 'ILLUMINA' ... 'ILLUMINA'
ReleaseDate (Sample) object '2016-01-04' ... '2016-01-04'
SRA_Study (Sample) object 'SRP065945' 'SRP065945' ... 'SRP065945'
source_name (Sample) object 'Rice leaf' 'Rice leaf' ... 'Rice leaf'
tissue (Sample) object 'leaf' 'leaf' 'leaf' ... 'leaf' 'leaf'
counts (Sample, Gene) int64 20 0 0 0 0 0 ... 0 52 335 0 666 0- Sample: 475
- Gene: 55986
- Sample(Sample)object'SRX1423934' ... 'SRX1424408'
array(['SRX1423934', 'SRX1423935', 'SRX1423936', ..., 'SRX1424406', 'SRX1424407', 'SRX1424408'], dtype=object) - Gene(Gene)object'LOC_Os06g05820' ... 'LOC_Os07g0...
array(['LOC_Os06g05820', 'LOC_Os10g27460', 'LOC_Os02g35980', ..., 'LOC_Os03g50190', 'LOC_Os03g20020', 'LOC_Os07g03418'], dtype=object)
- BioSample(Sample)object'SAMN04251848' ... 'SAMN04251607'
array(['SAMN04251848', 'SAMN04251849', 'SAMN04251850', 'SAMN04251851', 'SAMN04251852', 'SAMN04251853', 'SAMN04251854', 'SAMN04251855', 'SAMN04251856', 'SAMN04251857', 'SAMN04251858', 'SAMN04251859', 'SAMN04251860', 'SAMN04251861', 'SAMN04251862', 'SAMN04251863', 'SAMN04251864', 'SAMN04251865', 'SAMN04251866', 'SAMN04251867', 'SAMN04251868', 'SAMN04251869', 'SAMN04251870', 'SAMN04251871', 'SAMN04251872', 'SAMN04251873', 'SAMN04251874', 'SAMN04251875', 'SAMN04251876', 'SAMN04251877', 'SAMN04251878', 'SAMN04251879', 'SAMN04251880', 'SAMN04251881', 'SAMN04251882', 'SAMN04251883', 'SAMN04251884', 'SAMN04251885', 'SAMN04251886', 'SAMN04251887', 'SAMN04251888', 'SAMN04251889', 'SAMN04251890', 'SAMN04251891', 'SAMN04251892', 'SAMN04251893', 'SAMN04251894', 'SAMN04251895', 'SAMN04251896', 'SAMN04251897', 'SAMN04251898', 'SAMN04251899', 'SAMN04251900', 'SAMN04251901', 'SAMN04251902', 'SAMN04251903', 'SAMN04251964', 'SAMN04251965', 'SAMN04251966', 'SAMN04251967', 'SAMN04251968', 'SAMN04251969', 'SAMN04251970', 'SAMN04251971', 'SAMN04251972', 'SAMN04251973', 'SAMN04251974', 'SAMN04251975', 'SAMN04251976', 'SAMN04251977', 'SAMN04251978', 'SAMN04251979', 'SAMN04251980', 'SAMN04251981', 'SAMN04251982', 'SAMN04251983', 'SAMN04251984', 'SAMN04251985', 'SAMN04251986', 'SAMN04251987', ... 'SAMN04251618', 'SAMN04251619', 'SAMN04251620', 'SAMN04251621', 'SAMN04251622', 'SAMN04251623', 'SAMN04251624', 'SAMN04251625', 'SAMN04251626', 'SAMN04251627', 'SAMN04251628', 'SAMN04251629', 'SAMN04251630', 'SAMN04251631', 'SAMN04251632', 'SAMN04251633', 'SAMN04251634', 'SAMN04251635', 'SAMN04251636', 'SAMN04251637', 'SAMN04251549', 'SAMN04251550', 'SAMN04251551', 'SAMN04251552', 'SAMN04251553', 'SAMN04251554', 'SAMN04251555', 'SAMN04251556', 'SAMN04251557', 'SAMN04251558', 'SAMN04251559', 'SAMN04251560', 'SAMN04251561', 'SAMN04251562', 'SAMN04251563', 'SAMN04251564', 'SAMN04251565', 'SAMN04251566', 'SAMN04251567', 'SAMN04251568', 'SAMN04251569', 'SAMN04251570', 'SAMN04251571', 'SAMN04251572', 'SAMN04251573', 'SAMN04251574', 'SAMN04251575', 'SAMN04251576', 'SAMN04251577', 'SAMN04251578', 'SAMN04251579', 'SAMN04251580', 'SAMN04251581', 'SAMN04251582', 'SAMN04251583', 'SAMN04251584', 'SAMN04251585', 'SAMN04251586', 'SAMN04251587', 'SAMN04251588', 'SAMN04251589', 'SAMN04251590', 'SAMN04251591', 'SAMN04251592', 'SAMN04251593', 'SAMN04251594', 'SAMN04251595', 'SAMN04251596', 'SAMN04251597', 'SAMN04251598', 'SAMN04251599', 'SAMN04251600', 'SAMN04251601', 'SAMN04251602', 'SAMN04251603', 'SAMN04251604', 'SAMN04251605', 'SAMN04251606', 'SAMN04251607'], dtype=object) - LoadDate(Sample)object'2015-11-20' ... '2015-11-19'
array(['2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', '2015-11-20', ... '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19', '2015-11-19'], dtype=object) - MBases(Sample)int644016 5202 4053 ... 3098 3529 2922
array([ 4016, 5202, 4053, 1166, 4005, 4852, 3610, 6065, 3319, 3965, 3607, 1322, 2718, 3203, 4188, 1077, 2318, 3925, 3242, 3697, 4360, 3241, 3508, 3653, 4255, 3412, 3791, 3037, 2815, 3328, 4301, 4030, 4574, 3266, 2853, 3097, 3106, 5413, 2570, 3482, 1679, 4650, 1782, 3085, 5362, 2069, 1173, 3743, 5127, 2089, 2103, 3111, 2639, 3479, 3913, 3436, 4551, 1869, 4535, 3719, 2886, 3895, 4381, 3405, 3542, 1691, 4306, 3558, 3911, 3885, 3190, 4039, 3258, 3109, 5006, 4363, 3551, 3743, 3074, 3520, 3057, 3232, 4062, 3613, 4126, 4458, 1858, 2559, 4226, 2073, 1372, 4480, 3471, 4302, 3663, 3294, 5182, 2223, 1330, 3281, 2747, 4252, 3642, 3553, 2761, 4244, 3006, 3654, 3065, 14153, 3577, 2930, 3159, 3870, 3311, 4689, 4659, 4294, 3589, 3228, 3640, 4646, 3455, 2961, 4156, 3855, 4114, 3537, 3460, 3190, 4442, 3913, 3939, 2412, 2541, 2978, 2629, 3539, 3620, 3174, 3910, 2413, 2999, 3194, 3556, 3339, 3615, 2645, 3687, 3095, 4071, 3749, 2413, 2635, 4302, 2625, 3221, 2512, 4604, 3779, 2650, 3412, 4681, 3099, 3893, 2615, 2728, 2578, 3859, 5066, 3986, 3086, 3251, 2847, 3434, 3305, 2927, 3603, 3990, 3463, ... 3898, 2947, 3147, 4680, 3533, 2648, 2917, 3873, 2582, 3666, 2844, 4735, 2454, 2843, 4071, 4883, 3754, 2454, 3280, 5604, 1229, 3921, 3225, 3874, 3524, 3287, 3351, 2550, 3802, 2957, 4992, 3572, 2886, 2403, 3921, 3377, 3256, 2685, 3398, 2708, 3045, 2465, 3733, 4366, 3426, 3307, 4461, 3453, 2931, 4296, 3474, 3940, 3525, 3107, 3157, 3453, 5316, 3896, 3199, 4203, 4325, 3989, 2820, 4048, 2785, 4108, 3406, 3648, 3341, 4907, 3499, 3757, 2650, 3778, 4314, 3090, 4836, 3288, 3231, 3495, 3049, 3218, 4291, 3335, 4792, 2652, 5257, 4005, 2967, 4688, 3925, 2372, 4031, 3165, 3422, 3365, 1527, 3342, 3285, 3524, 2205, 1940, 3973, 3890, 1789, 1878, 3648, 4102, 1650, 1997, 2533, 4541, 3172, 4776, 3564, 2639, 3532, 3766, 3878, 2952, 4163, 3691, 3027, 3126, 3719, 4637, 3075, 3632, 3376, 4565, 3102, 3173, 3164, 4351, 3575, 3610, 4647, 4687, 3331, 2743, 2548, 3788, 3552, 942, 3825, 3099, 3343, 3075, 1360, 3574, 3290, 3196, 3032, 1743, 1431, 1672, 5345, 3209, 3784, 2975, 3206, 3576, 2971, 4036, 3153, 2413, 3643, 4206, 4572, 3214, 3566, 3025, 4099, 4177, 3648, 3098, 3529, 2922]) - MBytes(Sample)int642738 3652 2719 ... 1983 2370 1862
array([2738, 3652, 2719, 764, 2500, 3262, 2437, 4013, 2186, 2680, 2368, 877, 1783, 2105, 2772, 723, 1486, 2733, 2249, 2372, 2859, 2144, 2366, 2297, 2928, 2165, 2628, 1916, 1892, 2294, 2767, 2566, 2961, 2045, 1820, 1956, 1922, 3400, 1605, 2428, 1123, 3021, 1159, 2053, 3636, 1344, 768, 2434, 3454, 1315, 1314, 2043, 1765, 2251, 2453, 2227, 3068, 1210, 2966, 2369, 1880, 2417, 2969, 2195, 2208, 1059, 2904, 2239, 2549, 2699, 2060, 2554, 2065, 2014, 3340, 2783, 2405, 2472, 1981, 2274, 1967, 2057, 2664, 2244, 2654, 2908, 1220, 1640, 2705, 1297, 891, 3139, 2378, 2830, 2388, 2123, 3412, 1401, 837, 2103, 1866, 2740, 2366, 2267, 1818, 2933, 2005, 2309, 1945, 9778, 2292, 2025, 2033, 2520, 2108, 3025, 3066, 2912, 2429, 2080, 2452, 3105, 2216, 1879, 2762, 2558, 2650, 2316, 2337, 2161, 2881, 2538, 2726, 1691, 1658, 1877, 1664, 2292, 2330, 2046, 2672, 1676, 1956, 2004, 2272, 2082, 2515, 1683, 2531, 2029, 2625, 2495, 1550, 1771, 2797, 1650, 2040, 1607, 3202, 2405, 1698, 2177, 3144, 1994, 2544, 1674, 1824, 1656, 2554, 3317, 2658, 1927, 2010, 1780, 2197, 2212, 1868, 2301, 2730, 2211, 2518, 2087, 2291, 2453, 1901, 1883, 2186, 3265, 3497, 1542, 2356, 2271, 2659, 1516, 2006, 1856, 3006, 2611, 1669, 2282, 2347, 2745, 2017, 1853, 2439, 2133, 1305, 1748, 2221, 2533, 2065, 2802, 2317, 2345, 1724, 2047, 2223, 2525, 2222, 2586, ... 2656, 2823, 2021, 2677, 2069, 2432, 1245, 1469, 4581, 2386, 1891, 2101, 2657, 2068, 2205, 2682, 3083, 1887, 1580, 2397, 2365, 2666, 2340, 2505, 2101, 2892, 1783, 2927, 2986, 1479, 2657, 1950, 2486, 2537, 1995, 2011, 3146, 2427, 1692, 1825, 2605, 1650, 2357, 1807, 3255, 1574, 1903, 2716, 3170, 2531, 1544, 2113, 3774, 805, 2539, 2018, 2469, 2479, 2145, 2321, 1583, 2632, 1873, 3448, 2263, 1799, 1539, 2547, 2155, 2046, 1749, 2346, 1741, 1957, 1559, 2552, 2995, 2226, 2292, 3020, 2262, 1984, 2802, 2213, 2514, 2253, 2017, 2147, 2159, 3681, 2536, 2046, 2746, 2902, 2661, 1816, 2602, 1823, 2701, 2162, 2478, 2175, 3235, 2373, 2442, 1739, 2473, 2922, 2120, 3368, 2185, 2078, 2212, 1960, 2127, 2760, 2124, 3227, 1694, 3550, 2703, 2030, 3270, 2627, 1543, 2588, 2069, 2278, 2181, 1001, 2098, 2099, 2236, 1392, 1215, 2675, 2456, 1114, 1161, 2282, 2640, 1023, 1253, 1617, 2905, 2035, 3134, 2402, 1685, 2283, 2403, 2628, 1906, 2863, 2345, 1917, 1952, 2404, 3084, 2005, 2372, 2160, 3103, 1951, 2047, 2106, 2920, 2435, 2274, 3168, 3091, 2113, 1767, 1583, 2426, 2269, 624, 2504, 2033, 2144, 1923, 912, 2366, 2149, 2196, 2034, 1155, 958, 1153, 3596, 2034, 2599, 1908, 2063, 2372, 1900, 2734, 2133, 1634, 2432, 2693, 2942, 2084, 2313, 1940, 2736, 2865, 2505, 1983, 2370, 1862]) - Run(Sample)object'SRR2931040' ... 'SRR2931514'
array(['SRR2931040', 'SRR2931041', 'SRR2931042', 'SRR2931043', 'SRR2931044', 'SRR2931045', 'SRR2931046', 'SRR2931047', 'SRR2931048', 'SRR2931049', 'SRR2931050', 'SRR2931051', 'SRR2931052', 'SRR2931053', 'SRR2931054', 'SRR2931055', 'SRR2931056', 'SRR2931057', 'SRR2931058', 'SRR2931059', 'SRR2931060', 'SRR2931061', 'SRR2931062', 'SRR2931063', 'SRR2931064', 'SRR2931065', 'SRR2931066', 'SRR2931067', 'SRR2931068', 'SRR2931069', 'SRR2931070', 'SRR2931071', 'SRR2931072', 'SRR2931073', 'SRR2931074', 'SRR2931075', 'SRR2931076', 'SRR2931077', 'SRR2931078', 'SRR2931079', 'SRR2931080', 'SRR2931081', 'SRR2931082', 'SRR2931083', 'SRR2931084', 'SRR2931085', 'SRR2931086', 'SRR2931087', 'SRR2931088', 'SRR2931089', 'SRR2931090', 'SRR2931091', 'SRR2931092', 'SRR2931093', 'SRR2931094', 'SRR2931095', 'SRR2931096', 'SRR2931097', 'SRR2931098', 'SRR2931099', 'SRR2931100', 'SRR2931101', 'SRR2931102', 'SRR2931103', 'SRR2931104', 'SRR2931105', 'SRR2931106', 'SRR2931107', 'SRR2931108', 'SRR2931109', 'SRR2931110', 'SRR2931111', 'SRR2931112', 'SRR2931113', 'SRR2931114', 'SRR2931115', 'SRR2931116', 'SRR2931117', 'SRR2931118', 'SRR2931119', ... 'SRR2931436', 'SRR2931437', 'SRR2931438', 'SRR2931439', 'SRR2931440', 'SRR2931441', 'SRR2931442', 'SRR2931443', 'SRR2931444', 'SRR2931445', 'SRR2931446', 'SRR2931447', 'SRR2931448', 'SRR2931449', 'SRR2931450', 'SRR2931451', 'SRR2931452', 'SRR2931453', 'SRR2931454', 'SRR2931455', 'SRR2931456', 'SRR2931457', 'SRR2931458', 'SRR2931459', 'SRR2931460', 'SRR2931461', 'SRR2931462', 'SRR2931463', 'SRR2931464', 'SRR2931465', 'SRR2931466', 'SRR2931467', 'SRR2931468', 'SRR2931469', 'SRR2931470', 'SRR2931471', 'SRR2931472', 'SRR2931473', 'SRR2931474', 'SRR2931475', 'SRR2931476', 'SRR2931477', 'SRR2931478', 'SRR2931479', 'SRR2931480', 'SRR2931481', 'SRR2931482', 'SRR2931483', 'SRR2931484', 'SRR2931485', 'SRR2931486', 'SRR2931487', 'SRR2931488', 'SRR2931489', 'SRR2931490', 'SRR2931491', 'SRR2931492', 'SRR2931493', 'SRR2931494', 'SRR2931495', 'SRR2931496', 'SRR2931497', 'SRR2931498', 'SRR2931499', 'SRR2931500', 'SRR2931501', 'SRR2931502', 'SRR2931503', 'SRR2931504', 'SRR2931505', 'SRR2931506', 'SRR2931507', 'SRR2931508', 'SRR2931509', 'SRR2931510', 'SRR2931511', 'SRR2931512', 'SRR2931513', 'SRR2931514'], dtype=object) - SRA_Sample(Sample)object'SRS1156722' ... 'SRS1156251'
array(['SRS1156722', 'SRS1156721', 'SRS1156718', 'SRS1156717', 'SRS1156720', 'SRS1156719', 'SRS1156716', 'SRS1156714', 'SRS1156715', 'SRS1156713', 'SRS1156711', 'SRS1156712', 'SRS1156710', 'SRS1156709', 'SRS1156708', 'SRS1156707', 'SRS1156706', 'SRS1156705', 'SRS1156703', 'SRS1156704', 'SRS1156702', 'SRS1156700', 'SRS1156698', 'SRS1156699', 'SRS1156701', 'SRS1156696', 'SRS1156697', 'SRS1156695', 'SRS1156694', 'SRS1156693', 'SRS1156691', 'SRS1156692', 'SRS1156690', 'SRS1156688', 'SRS1156689', 'SRS1156687', 'SRS1156686', 'SRS1156684', 'SRS1156685', 'SRS1156683', 'SRS1156682', 'SRS1156681', 'SRS1156680', 'SRS1156678', 'SRS1156679', 'SRS1156677', 'SRS1156676', 'SRS1156675', 'SRS1156674', 'SRS1156673', 'SRS1156672', 'SRS1156671', 'SRS1156668', 'SRS1156669', 'SRS1156670', 'SRS1156667', 'SRS1156666', 'SRS1156664', 'SRS1156665', 'SRS1156663', 'SRS1156662', 'SRS1156659', 'SRS1156660', 'SRS1156661', 'SRS1156658', 'SRS1156657', 'SRS1156656', 'SRS1156655', 'SRS1156654', 'SRS1156653', 'SRS1156652', 'SRS1156651', 'SRS1156650', 'SRS1156649', 'SRS1156648', 'SRS1156647', 'SRS1156646', 'SRS1156645', 'SRS1156644', 'SRS1156643', ... 'SRS1156328', 'SRS1156327', 'SRS1156326', 'SRS1156325', 'SRS1156324', 'SRS1156323', 'SRS1156321', 'SRS1156322', 'SRS1156320', 'SRS1156319', 'SRS1156318', 'SRS1156317', 'SRS1156316', 'SRS1156315', 'SRS1156314', 'SRS1156313', 'SRS1156312', 'SRS1156311', 'SRS1156309', 'SRS1156310', 'SRS1156308', 'SRS1156307', 'SRS1156306', 'SRS1156305', 'SRS1156304', 'SRS1156303', 'SRS1156302', 'SRS1156301', 'SRS1156299', 'SRS1156300', 'SRS1156298', 'SRS1156297', 'SRS1156296', 'SRS1156295', 'SRS1156294', 'SRS1156292', 'SRS1156293', 'SRS1156249', 'SRS1156291', 'SRS1156290', 'SRS1156289', 'SRS1156288', 'SRS1156287', 'SRS1156286', 'SRS1156285', 'SRS1156284', 'SRS1156282', 'SRS1156283', 'SRS1156281', 'SRS1156280', 'SRS1156279', 'SRS1156277', 'SRS1156278', 'SRS1156276', 'SRS1156275', 'SRS1156274', 'SRS1156273', 'SRS1156272', 'SRS1156271', 'SRS1156270', 'SRS1156269', 'SRS1156268', 'SRS1156267', 'SRS1156266', 'SRS1156250', 'SRS1156265', 'SRS1156264', 'SRS1156263', 'SRS1156262', 'SRS1156261', 'SRS1156260', 'SRS1156258', 'SRS1156259', 'SRS1156257', 'SRS1156256', 'SRS1156255', 'SRS1156254', 'SRS1156252', 'SRS1156251'], dtype=object) - Sample_Name(Sample)object'GSM1933346' ... 'GSM1933820'
array(['GSM1933346', 'GSM1933347', 'GSM1933348', 'GSM1933349', 'GSM1933350', 'GSM1933351', 'GSM1933352', 'GSM1933353', 'GSM1933354', 'GSM1933355', 'GSM1933356', 'GSM1933357', 'GSM1933358', 'GSM1933359', 'GSM1933360', 'GSM1933361', 'GSM1933362', 'GSM1933363', 'GSM1933364', 'GSM1933365', 'GSM1933366', 'GSM1933367', 'GSM1933368', 'GSM1933369', 'GSM1933370', 'GSM1933371', 'GSM1933372', 'GSM1933373', 'GSM1933374', 'GSM1933375', 'GSM1933376', 'GSM1933377', 'GSM1933378', 'GSM1933379', 'GSM1933380', 'GSM1933381', 'GSM1933382', 'GSM1933383', 'GSM1933384', 'GSM1933385', 'GSM1933386', 'GSM1933387', 'GSM1933388', 'GSM1933389', 'GSM1933390', 'GSM1933391', 'GSM1933392', 'GSM1933393', 'GSM1933394', 'GSM1933395', 'GSM1933396', 'GSM1933397', 'GSM1933398', 'GSM1933399', 'GSM1933400', 'GSM1933401', 'GSM1933402', 'GSM1933403', 'GSM1933404', 'GSM1933405', 'GSM1933406', 'GSM1933407', 'GSM1933408', 'GSM1933409', 'GSM1933410', 'GSM1933411', 'GSM1933412', 'GSM1933413', 'GSM1933414', 'GSM1933415', 'GSM1933416', 'GSM1933417', 'GSM1933418', 'GSM1933419', 'GSM1933420', 'GSM1933421', 'GSM1933422', 'GSM1933423', 'GSM1933424', 'GSM1933425', ... 'GSM1933742', 'GSM1933743', 'GSM1933744', 'GSM1933745', 'GSM1933746', 'GSM1933747', 'GSM1933748', 'GSM1933749', 'GSM1933750', 'GSM1933751', 'GSM1933752', 'GSM1933753', 'GSM1933754', 'GSM1933755', 'GSM1933756', 'GSM1933757', 'GSM1933758', 'GSM1933759', 'GSM1933760', 'GSM1933761', 'GSM1933762', 'GSM1933763', 'GSM1933764', 'GSM1933765', 'GSM1933766', 'GSM1933767', 'GSM1933768', 'GSM1933769', 'GSM1933770', 'GSM1933771', 'GSM1933772', 'GSM1933773', 'GSM1933774', 'GSM1933775', 'GSM1933776', 'GSM1933777', 'GSM1933778', 'GSM1933779', 'GSM1933780', 'GSM1933781', 'GSM1933782', 'GSM1933783', 'GSM1933784', 'GSM1933785', 'GSM1933786', 'GSM1933787', 'GSM1933788', 'GSM1933789', 'GSM1933790', 'GSM1933791', 'GSM1933792', 'GSM1933793', 'GSM1933794', 'GSM1933795', 'GSM1933796', 'GSM1933797', 'GSM1933798', 'GSM1933799', 'GSM1933800', 'GSM1933801', 'GSM1933802', 'GSM1933803', 'GSM1933804', 'GSM1933805', 'GSM1933806', 'GSM1933807', 'GSM1933808', 'GSM1933809', 'GSM1933810', 'GSM1933811', 'GSM1933812', 'GSM1933813', 'GSM1933814', 'GSM1933815', 'GSM1933816', 'GSM1933817', 'GSM1933818', 'GSM1933819', 'GSM1933820'], dtype=object) - genotype(Sample)object'Azuenca (AZ; IRGC#328, Japonica...
array(['Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', 'Azuenca (AZ; IRGC#328, Japonica)', ... 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)', 'Tadukan (TD; IRGC#9804, Indica)'], dtype=object) - time(Sample)int6415 15 30 30 45 ... 270 270 300 300
array([ 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 225, 240, 270, 270, 300, 300, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 105, 105, 120, 120, 150, 150, 165, 165, 180, 180, 210, 210, 240, 240, 270, 270, 300, 300, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 270, 270, 300, 300, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 105, 105, 120, 120, 135, 135, 105, 105, 120, 120, 150, 150, 165, 165, 180, 180, 210, 210, 240, 240, 270, 270, 300, 300, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 270, 270, 300, 300, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 105, 120, 120, 150, 150, 165, 165, 180, 180, 210, 210, 240, 240, 270, 270, 300, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 270, 270, 300, 300, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 135, 135, 150, 150, 165, 165, 180, 180, 195, 195, 210, 210, 225, 225, 240, 240, 15, 15, 30, 30, 45, 45, 60, 60, 75, 75, 90, 90, 105, 105, 120, 120, 135, 135, 105, 105, 120, 120, 150, 150, 165, 165, 180, 180, 210, 210, 240, 240, 270, 270, 300, 300]) - treatment(Sample)object'CONTROL' ... 'RECOV_DROUGHT'
array(['CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', ... 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'CONTROL', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'RECOV_HEAT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT', 'RECOV_DROUGHT'], dtype=object) - Assay_Type(Sample)object'RNA-Seq' 'RNA-Seq' ... 'RNA-Seq'
array(['RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', ... 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq', 'RNA-Seq'], dtype=object) - AvgSpotLen(Sample)int64102 102 102 102 ... 102 102 102 102
array([102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102, 102]) - BioProject(Sample)object'PRJNA301554' ... 'PRJNA301554'
array(['PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', ... 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554', 'PRJNA301554'], dtype=object) - Center_Name(Sample)object'GEO' 'GEO' 'GEO' ... 'GEO' 'GEO'
array(['GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', ... 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO', 'GEO'], dtype=object) - Consent(Sample)object'public' 'public' ... 'public'
array(['public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', ... 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public', 'public'], dtype=object) - DATASTORE_filetype(Sample)object'sra' 'sra' 'sra' ... 'sra' 'sra'
array(['sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', ... 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra', 'sra'], dtype=object) - DATASTORE_provider(Sample)object'ncbi' 'ncbi' ... 'ncbi' 'ncbi'
array(['ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', ... 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi', 'ncbi'], dtype=object) - InsertSize(Sample)int640 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0
array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]) - Instrument(Sample)object'Illumina HiSeq 2000' ... 'Illum...
array(['Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', ... 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000', 'Illumina HiSeq 2000'], dtype=object) - LibraryLayout(Sample)object'PAIRED' 'PAIRED' ... 'PAIRED'
array(['PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', ... 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED', 'PAIRED'], dtype=object) - LibrarySelection(Sample)object'cDNA' 'cDNA' ... 'cDNA' 'cDNA'
array(['cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', ... 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA', 'cDNA'], dtype=object) - LibrarySource(Sample)object'TRANSCRIPTOMIC' ... 'TRANSCRIPT...
array(['TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', ... 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC', 'TRANSCRIPTOMIC'], dtype=object) - Organism(Sample)object'Oryza sativa' ... 'Oryza sativa'
array(['Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', ... 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa', 'Oryza sativa'], dtype=object) - Platform(Sample)object'ILLUMINA' ... 'ILLUMINA'
array(['ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', ... 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA', 'ILLUMINA'], dtype=object) - ReleaseDate(Sample)object'2016-01-04' ... '2016-01-04'
array(['2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', ... '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04', '2016-01-04'], dtype=object) - SRA_Study(Sample)object'SRP065945' ... 'SRP065945'
array(['SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', ... 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945', 'SRP065945'], dtype=object) - source_name(Sample)object'Rice leaf' ... 'Rice leaf'
array(['Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', ... 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf', 'Rice leaf'], dtype=object) - tissue(Sample)object'leaf' 'leaf' ... 'leaf' 'leaf'
array(['leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', ... 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf', 'leaf'], dtype=object) - counts(Sample, Gene)int6420 0 0 0 0 0 ... 0 52 335 0 666 0
array([[ 20, 0, 0, ..., 0, 637, 0], [ 2, 0, 0, ..., 0, 186, 0], [ 22, 0, 0, ..., 0, 545, 0], ..., [ 8, 0, 0, ..., 0, 411, 0], [ 8, 0, 0, ..., 0, 311, 0], [ 21, 0, 0, ..., 0, 666, 0]])
Add a Normalized or Transformed Count Matrix¶
The AnnotatedGEM object can hold more than one count matrix, so long as they share the same gene and sample
coordinates. Here we demonstrate adding a TPM normalized matrix as produced by edgeR. This is more useful for
transforms that are computationally expensive, or that require data not easily stored in the AnnotatedGEM object.
We can then access a given count matrix by passing count_variable='NAME' to get_gem_data().
counts, _ = gsf.get_gem_data(agem)
agem.data['qt_counts'] = xr.DataArray(
quantile_transform(counts.values, output_distribution='normal', axis=1),
coords=counts.coords,
name='qt_counts')
The xarray.DataSet object is available as .data:
agem.data['qt_counts']
<xarray.DataArray 'qt_counts' (Sample: 475, Gene: 55986)>
array([[ 0.52930109, -5.19933758, -5.19933758, ..., -5.19933758,
1.59769842, -5.19933758],
[ 0.36687305, -5.19933758, -5.19933758, ..., -5.19933758,
1.54592732, -5.19933758],
[ 0.53074486, -5.19933758, -5.19933758, ..., -5.19933758,
1.53361204, -5.19933758],
...,
[ 0.4279759 , -5.19933758, -5.19933758, ..., -5.19933758,
1.51246295, -5.19933758],
[ 0.42248277, -5.19933758, -5.19933758, ..., -5.19933758,
1.36271302, -5.19933758],
[ 0.5642668 , -5.19933758, -5.19933758, ..., -5.19933758,
1.74700661, -5.19933758]])
Coordinates:
* Gene (Gene) object 'LOC_Os06g05820' ... 'LOC_Os07g03418'
* Sample (Sample) object 'SRX1423934' 'SRX1423935' ... 'SRX1424408'- Sample: 475
- Gene: 55986
- 0.5293 -5.199 -5.199 -5.199 -5.199 ... 1.415 -5.199 1.747 -5.199
array([[ 0.52930109, -5.19933758, -5.19933758, ..., -5.19933758, 1.59769842, -5.19933758], [ 0.36687305, -5.19933758, -5.19933758, ..., -5.19933758, 1.54592732, -5.19933758], [ 0.53074486, -5.19933758, -5.19933758, ..., -5.19933758, 1.53361204, -5.19933758], ..., [ 0.4279759 , -5.19933758, -5.19933758, ..., -5.19933758, 1.51246295, -5.19933758], [ 0.42248277, -5.19933758, -5.19933758, ..., -5.19933758, 1.36271302, -5.19933758], [ 0.5642668 , -5.19933758, -5.19933758, ..., -5.19933758, 1.74700661, -5.19933758]]) - Gene(Gene)object'LOC_Os06g05820' ... 'LOC_Os07g0...
array(['LOC_Os06g05820', 'LOC_Os10g27460', 'LOC_Os02g35980', ..., 'LOC_Os03g50190', 'LOC_Os03g20020', 'LOC_Os07g03418'], dtype=object) - Sample(Sample)object'SRX1423934' ... 'SRX1424408'
array(['SRX1423934', 'SRX1423935', 'SRX1423936', ..., 'SRX1424406', 'SRX1424407', 'SRX1424408'], dtype=object)
Select Counts and Annotations using get_gem_data()¶
The AnnotatedGEM object (and the GeneSetCollection, introduced further down) can have data subsets pulled
from them easily using the get_gem_data() interface. Here we select and pass genes to
UMAP, a dimensional reduction technique we prefer over
PCA, tSNE and others.
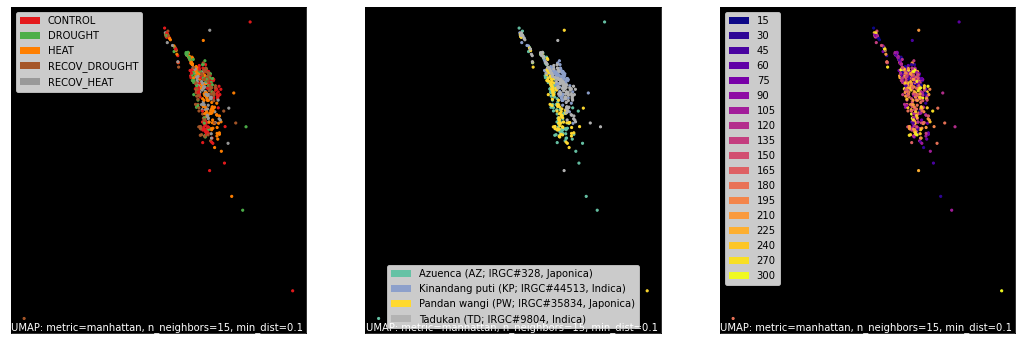
counts, labels = gsf.get_gem_data(agem, annotation_variables=['treatment', 'genotype', 'time'])
mapper = umap.UMAP(densmap=True, random_state=42, metric='manhattan').fit(counts.values)
fig, axes = plt.subplots(1, 3, figsize=(18, 6))
umap.plot.points(mapper, labels=labels['treatment'], background='black', ax=axes[0], color_key_cmap='Set1');
umap.plot.points(mapper, labels=labels['genotype'], background='black', ax=axes[1], color_key_cmap='Set2');
umap.plot.points(mapper, labels=labels['time'], background='black', ax=axes[2], color_key_cmap='plasma');
counts, labels = gsf.get_gem_data(agem, annotation_variables=['treatment', 'genotype', 'time'], count_variable='qt_counts')
mapper = umap.UMAP(densmap=True, random_state=42, metric='manhattan').fit(counts.values)
fig, axes = plt.subplots(1, 3, figsize=(18, 6))
umap.plot.points(mapper, labels=labels['treatment'], background='black', ax=axes[0], color_key_cmap='Set1');
umap.plot.points(mapper, labels=labels['genotype'], background='black', ax=axes[1], color_key_cmap='Set2');
umap.plot.points(mapper, labels=labels['time'], background='black', ax=axes[2], color_key_cmap='plasma');

2. Select genes / features / tags¶
While we can use the formula interface in R, it is often desirable to re-use the same design matrix and contrasts
across feature selection methods. We use the patsy package to
accomplish this.
I end up creating multiple `GeneSetCollection` objects here, while I could have just as easily used one.
How many collection objects used depends on your preferences and the complexity of your analysis.
Create Design Matrices and Contrasts¶
Here I make some simple contrasts that select for treatment from each of our designs.
Newer users may find it easier to use the
makeContrasts function
provided by the popular limma package. I create ordered, nested lists for easier transfer to and from R.
counts, labels = gsf.get_gem_data(agem, annotation_variables=['treatment', 'time', 'genotype'])
labels = labels.to_dataframe()
ri_counts = gsf.utils.R_interface.Py_counts_to_R(counts)
forumla_designs = [
"0 + treatment",
"0 + treatment:genotype",
# These models take too long to run for a demo-notebook.
# "0 + treatment:genotype:scale(time, center=False)",
# "0 + treatment:genotype:C(time):C(replicate)",
]
# I use nested lists instead of dictionaries to facilitate transfering to and from R.
design_list = [[f, patsy.dmatrix(f, labels)] for f in forumla_designs]
dmatrix_list = [[f, np.asarray(v)] for f, v in design_list]
def make_contrast(design, factors):
"""Constructs a contrast array for each item in factors. Returns a one-hot array."""
return np.sum([(np.core.defchararray.find(design.design_info.column_names, f)!=-1).astype(int)
for f in factors], axis=0)
treatment_factors = design_list[0][1].design_info.column_names[1:]
control_factor = design_list[0][1].design_info.column_names[0]
contrast_list = list()
for i, (formula, design) in enumerate(design_list):
design_contrast = list()
for j, col_name in enumerate(treatment_factors):
contrast = make_contrast(design, [col_name]) - make_contrast(design, [control_factor])
design_contrast.append([col_name, contrast])
contrast_list.append(design_contrast)
Let’s view the simplest model and the contrasts created:
dmatrix_list[0]
['0 + treatment',
array([[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
...,
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.]])]
contrast_list[0]
[['treatment[DROUGHT]', array([-1, 1, 0, 0, 0])],
['treatment[HEAT]', array([-1, 0, 1, 0, 0])],
['treatment[RECOV_DROUGHT]', array([-1, 0, 0, 1, 0])],
['treatment[RECOV_HEAT]', array([-1, 0, 0, 0, 1])]]
Select via edgeR¶
Here we present a ‘standard’ feature selection with edgeR, see the edgeR Bioconductor page for more information.
%%R -i ri_counts -i dmatrix_list -i contrast_list -o results -o keep
results <- list()
di <- 1
print('Applying filter.')
# Apply default gene filter.
keep <- filterByExpr(ri_counts, design=dmatrix_list[[1]][[2]])
Y <- ri_counts[keep,]
for (design in dmatrix_list)
{
name <- design[[1]]
design_matrix <- design[[2]]
print(sprintf('Examing design %s', name))
print('Construct the edgeR DGEList object.')
# Construct the edge list object.
dge_list <- DGEList(counts=Y)
dge_list <- calcNormFactors(dge_list)
dge_list <- estimateDisp(dge_list, design=design_matrix)
# Plot variation.
dir.create('disp_plots', showWarnings = FALSE)
png(sprintf("disp_plots/%s_counts_disp.png", name))
plotBCV(dge_list)
dev.off()
# Fit the negative binomial model.
fit <- glmQLFit(dge_list, design=design_matrix)
contrast_results <- list()
ci <- 1
for (contrast in contrast_list[[di]])
{
cname <- contrast[[1]]
contrast_array <- contrast[[2]]
print(sprintf('Examing contrast %s', cname))
contrast_result = glmQLFTest(fit, contrast=contrast_array)
p_values <- contrast_result$table$PValue
FDR <- p.adjust(p_values, method="BH")
contrast_result$table$FDR <- FDR
contrast_result$table$support_dir <- c(decideTests(contrast_result, p.value=0.001, lfc=2.0))
contrast_result$table$support <- abs(c(decideTests(contrast_result, p.value=0.001, lfc=2.0)))
contrast_results[[ ci ]] = contrast_result$table
ci <- ci + 1
}
results[[di]] <- contrast_results
di <- di + 1
}
[1] "Applying filter."
[1] "Examing design 0 + treatment"
[1] "Construct the edgeR DGEList object."
[1] "Examing contrast treatment[DROUGHT]"
[1] "Examing contrast treatment[HEAT]"
[1] "Examing contrast treatment[RECOV_DROUGHT]"
[1] "Examing contrast treatment[RECOV_HEAT]"
[1] "Examing design 0 + treatment:genotype"
[1] "Construct the edgeR DGEList object."
[1] "Examing contrast treatment[DROUGHT]"
[1] "Examing contrast treatment[HEAT]"
[1] "Examing contrast treatment[RECOV_DROUGHT]"
[1] "Examing contrast treatment[RECOV_HEAT]"
dge_collection = gsf.GeneSetCollection(gem=agem, name='DGE Results')
dge_collection['edgeR filter'] = gsf.GeneSet.from_bool_array(
np.asarray(keep).astype(bool),
agem.data.Gene.values,
name='edgeR filter')
for result_dfs, formula, contrasts in zip(results, forumla_designs, contrast_list):
for contrast_df, (contrast_name, _) in zip(list(result_dfs), contrasts):
key = f"'{formula}'__{contrast_name}"
dge_collection[key] = gsf.GeneSet(contrast_df, name=key)
dge_collection
<GSForge.GeneSetCollection>
DGE Results
GeneSets (9 total): Support Count
edgeR filter: 21915
'0 + treatment:genotype'__treatment[HEAT]: 4071
'0 + treatment:genotype'__treatment[RECOV_HEAT]: 3719
'0 + treatment:genotype'__treatment[DROUGHT]: 3703
'0 + treatment:genotype'__treatment[RECOV_DROUGHT]: 2806
... and 4 more.
dge_collection['edgeR filter']
<GSForge.GeneSet>
Name: edgeR filter
Supported Genes: 21915
gsf.plots.gem.RasterGEM(dge_collection, hue='genotype')

gsf.plots.gem.RasterGEM(dge_collection, selected_gene_sets=['edgeR filter'], hue='genotype')

if not TOUR_DGE.exists():
dge_collection.save(TOUR_DGE)
Random Forest via Boruta Feature Selection¶
See this FAQ for details on Boruta.
In brief, boruta lets us use a ‘minimal optimal’ feature selection model as an ‘all relevant’ method. Random forest
models typically work by finding a minimum optimal set of features. In our use, we are interested in all features
that may be relevant, not the minimum required to train a model. GSForge provides a wrapper which returns
xarray.DataSet objects suitable for immediate GeneSet construction.
boruta_gsc = gsf.GeneSetCollection(gem=agem, name='Boruta Results')
selecting_model = RandomForestClassifier(
class_weight='balanced',
max_depth=4,
n_estimators=100,
n_jobs=-1)
for target in ["treatment", "genotype"]:
boruta_treatment_ds = gsf.operations.BorutaProspector(
agem,
estimator=selecting_model,
annotation_variables=target,
max_iter=50)
boruta_gsc[f"Boruta_{target}"] = gsf.GeneSet(boruta_treatment_ds, name=f"Boruta_{target}")
boruta_gsc
<GSForge.GeneSetCollection>
Boruta Results
GeneSets (2 total): Support Count
Boruta_treatment: 757
Boruta_genotype: 662
if not TOUR_BORUTA.exists():
boruta_gsc.save(TOUR_BORUTA)
We can check if boruta still has features to resolve into important or not by examining the ‘support_weak’ variable. In practice one should increase the number of iterations until there are no remaining values to be resolved.
boruta_gsc['Boruta_genotype'].data.support_weak.values.sum()
38
Gene Sets from Literature¶
In practice there is often a gene set that is externally defined that we wish to compare. Fortunately the only requirement for a GeneSet to be useful is that has an index that matches that in our GEM. See this notebook for more details.
Here we read in a supporting information file from the EGRIN study by Wilkins et. al., and after a bit of parsing it is converted to a series of GeneSet objects.
This SI table appears to be counts of significant differential expression for time contrasts within each treatment.
The full model is not (exactly) specified in the paper, but we can infer that it was probably:
Where \(C(\text{label})\) indicates treatment as a categorical, rather than scalar variable. The union for each set of treatment contrasts was probably then used.
si1_df = pd.read_csv(SI_FILE_1_PATH, skiprows=3, index_col=0)
lit_dge_coll = gsf.GeneSetCollection(gem=agem, name='Literature DGE')
for col in si1_df.columns:
genes = si1_df[si1_df[col] > 0].index.values
diff = np.setdiff1d(genes, agem.data.Gene.values)
if diff.shape[0] > 0:
print(diff)
lit_dge_coll[col] = gsf.GeneSet.from_gene_array(genes, name=col)
lit_dge_coll.summarize_gene_sets()
{'DROUGHT_UP': 1175,
'HEAT_UP': 592,
'RECOV_DROUGHT_UP': 446,
'DROUGHT_DOWN': 170,
'HEAT_DOWN': 106,
'RECOV_HEAT_UP': 76,
'RECOV_DROUGHT_DOWN': 58,
'RECOV_HEAT_DOWN': 43}
3. Compare Results¶
While this step is ultimately experiment-specific, it usually entails some combination of:
Comparing selected to unselected features by one or more measure.
p-values and log-fold-change.
Ranking or comparing within a selected set.
Random forest feature importance.
p-values and log-fold-change.
Comparing set memberships, these usually take the form of set operations, such as:
union
intersection
difference
unique
Comparing between selection sets.
Model prediction scores.
Entries 1 and 2 are typically routine within the same selection set, as the tool used to create that set should provide the needed measures. More complications emerge with steps 3 and 4, especially when comparing selection sets that derive from different methods. Without additional wet lab experimentation we are limited in declaring our success in feature identification. Instead we can score our selected subset as it preforms in prediction models.
For this demonstration we will combine each collection into its own set by taking the union of their selections. Then we will examine the features selected for all treatments by each method.
union_coll = gsf.GeneSetCollection(gem=agem, name='Combnied Collection')
union_coll.gene_sets.update(boruta_gsc.gene_sets)
lit_geneset = gsf.GeneSet.from_GeneSets(*lit_dge_coll.gene_sets.values(), name='literature_union')
union_coll.gene_sets.update({'literature_union': lit_geneset})
# Here I get the keys for each of the two DGE models we ran above.
dge_keys = pd.Series(dge_collection.gene_sets.keys())
key_sets = [dge_keys[dge_keys.str.startswith(f"'{f}'")].values for f in forumla_designs]
for f, keys in zip(forumla_designs, key_sets):
name = f"combined '{f}'"
union_coll[name] = gsf.GeneSet.from_GeneSets(
*[dge_collection[k] for k in keys],
name=name)
union_coll
<GSForge.GeneSetCollection>
Combnied Collection
GeneSets (5 total): Support Count
combined '0 + treatment:genotype': 8941
literature_union: 2061
Boruta_treatment: 757
Boruta_genotype: 662
combined '0 + treatment': 596
Visualize Set Overlap¶
See the upsetplot documentation for more details.
Instead of a Venn diagram we use an ‘Upset plot’. This allows us to view overlaps of sets larger than three.
gsf.plots.collections.UpsetPlotInterface(union_coll)
<upsetplot.plotting.UpSet at 0x7fc3381b3850>
Comparing Selection Sets¶
We can estimate how well a given subset of genes ‘describes’ a sample (phenotype) label by comparing how well they perform using a given machine learning model.
results = dict()
for key in list(union_coll.gene_sets.keys()) + ['all']:
counts, treatment = gsf.get_gem_data(union_coll, selected_gene_sets=[key], annotation_variables=["treatment"])
x_train, x_test, y_train, y_test = model_selection.train_test_split(counts, treatment)
# model = RandomForestClassifier(class_weight='balanced', n_estimators=1000, n_jobs=-1, max_depth=6)
model = linear_model.Perceptron()
model.fit(x_train, y_train)
results[key] = model.score(x_test, y_test)
hv.Bars(results, kdims=["Gene Selection Group"]).opts(
xrotation=90, invert_axes=True, ylim=(0, 1.1), ylabel='Score', fig_size=150,
aspect=2, title='Scores vs Treatment Labels')

Ranking Features within Sets¶
GSForge provides helper functions to extract genes by a score values. Note we get the data from the original dge collection, as those logFC values are intact.
dge_ds = dge_collection["'0 + treatment:genotype'__treatment[HEAT]"]
dge_ds.get_top_n_genes("logFC", 10)
array(['LOC_Os03g14180', 'LOC_Os11g13980', 'LOC_Os04g01740',
'LOC_Os01g04370', 'LOC_Os01g04360', 'LOC_Os07g47840',
'LOC_Os02g15930', 'LOC_Os01g04380', 'LOC_Os11g05170',
'LOC_Os04g36750'], dtype=object)
dge_ds.get_genes_by_threshold(3.0, "logFC")
array(['ChrSy.fgenesh.gene.85', 'ChrSy.fgenesh.gene.86', 'LOC_Os01g01170',
..., 'LOC_Os12g43640', 'LOC_Os12g43850', 'LOC_Os12g44250'],
dtype=object)
Rank Genes with a Random Forest¶
Random forests and feature ranks. Robust enough to function in our case. Some values filtered prior to dge analysis…
counts.coords.dims[1]
'Gene'
counts.coords[counts.coords.dims[1]].values
array(['ChrSy.fgenesh.gene.21', 'ChrSy.fgenesh.gene.25',
'ChrSy.fgenesh.gene.28', ..., 'LOC_Os12g44310', 'LOC_Os12g44340',
'LOC_Os12g44370'], dtype=object)
union_coll["'0 + treatment:genotype'__treatment[HEAT]"] = dge_collection["'0 + treatment:genotype'__treatment[HEAT]"]
gene_rank_mdl = RandomForestClassifier(class_weight='balanced', n_estimators=1000, n_jobs=-2)
treatment_nFDR = gsf.operations.nFDR(
union_coll,
selected_gene_sets=["Boruta_treatment", "'0 + treatment:genotype'__treatment[HEAT]"],
gene_set_mode="union",
annotation_variables=["treatment"],
model=gene_rank_mdl,
n_iterations=5
)
treatment_feature_importance = gsf.operations.RankGenesByModel(
union_coll,
selected_gene_sets=["Boruta_treatment", "'0 + treatment:genotype'__treatment[HEAT]"],
gene_set_mode="union",
annotation_variables=["treatment"],
model=gene_rank_mdl,
n_iterations=5
)
treatment_feature_importance
<xarray.DataArray 'feature_importances' (model_iteration: 5, Gene: 4391)>
array([[1.31563510e-04, 6.79631213e-05, 2.29502259e-04, ...,
8.92052648e-05, 1.45398837e-04, 7.24346971e-05],
[2.84511837e-04, 6.37763120e-05, 2.20714802e-04, ...,
1.33545198e-04, 6.46603741e-05, 5.46214076e-05],
[9.88199062e-05, 2.69261394e-05, 1.67553915e-04, ...,
5.66013391e-05, 1.20388075e-04, 3.19671445e-05],
[3.36803404e-04, 4.91265354e-05, 3.29251471e-04, ...,
5.84467514e-05, 1.42280408e-04, 5.73415577e-05],
[2.85515835e-04, 2.21027518e-05, 2.11691414e-04, ...,
7.19938839e-05, 3.35372519e-05, 4.97356344e-05]])
Coordinates:
* Gene (Gene) object 'ChrSy.fgenesh.gene.37' ... 'LOC_Os12g44250'
Dimensions without coordinates: model_iteration
Attributes:
Ranking Model: RandomForestClassifier(class_weight='balanced', n_...
count_variable: counts
annotation_variables: treatment- model_iteration: 5
- Gene: 4391
- 0.0001316 6.796e-05 0.0002295 ... 7.199e-05 3.354e-05 4.974e-05
array([[1.31563510e-04, 6.79631213e-05, 2.29502259e-04, ..., 8.92052648e-05, 1.45398837e-04, 7.24346971e-05], [2.84511837e-04, 6.37763120e-05, 2.20714802e-04, ..., 1.33545198e-04, 6.46603741e-05, 5.46214076e-05], [9.88199062e-05, 2.69261394e-05, 1.67553915e-04, ..., 5.66013391e-05, 1.20388075e-04, 3.19671445e-05], [3.36803404e-04, 4.91265354e-05, 3.29251471e-04, ..., 5.84467514e-05, 1.42280408e-04, 5.73415577e-05], [2.85515835e-04, 2.21027518e-05, 2.11691414e-04, ..., 7.19938839e-05, 3.35372519e-05, 4.97356344e-05]]) - Gene(Gene)object'ChrSy.fgenesh.gene.37' ... 'LOC...
array(['ChrSy.fgenesh.gene.37', 'ChrSy.fgenesh.gene.85', 'ChrSy.fgenesh.gene.86', ..., 'LOC_Os12g44100', 'LOC_Os12g44110', 'LOC_Os12g44250'], dtype=object)
- Ranking Model :
- RandomForestClassifier(class_weight='balanced', n_estimators=1000, n_jobs=-2)
- count_variable :
- counts
- annotation_variables :
- treatment
gene_union = union_coll.union(["Boruta_treatment", "'0 + treatment:genotype'__treatment[HEAT]"])
boruta_support = np.isin(gene_union, union_coll["Boruta_treatment"].get_support())
dge_support = np.isin(gene_union,
union_coll["'0 + treatment:genotype'__treatment[HEAT]"].get_support())
support = np.zeros_like(gene_union)
support[boruta_support] = "Boruta"
support[dge_support] = "DGE"
support[(boruta_support * dge_support) == True] = "Both"
df = pd.DataFrame({
"logFC": dge_ds.data['logFC'].reindex(Gene=gene_union).values,
"PValue": dge_ds.data['PValue'].reindex(Gene=gene_union).values,
# "F": dge_ds.data['F'].reindex(Gene=gene_union).values,
"Gene": gene_union,
"mean feature importance": treatment_feature_importance.mean(dim="model_iteration").values,
"mean nFDR": treatment_nFDR.mean(dim="model_iteration").values,
"support source": support,
}).set_index("Gene")
# sns.pairplot(df, hue="support source", markers=['.', '.', 'X'],
# vars=["mean feature importance", "logFC", "mean nFDR"],
# plot_kws=dict(edgecolor=None, alpha=0.25));
fig, axes = plt.subplots(1, 3, figsize=(18, 6))
sns.scatterplot(data=df, x='mean feature importance', y='mean nFDR', hue='support source',
edgecolor=None, alpha=0.5, markers=['.', '.', 'X'], style='support source', ax=axes[0]);
sns.scatterplot(data=df, x='mean feature importance', y='logFC', hue='support source',
edgecolor=None, alpha=0.5, markers=['.', '.', 'X'], style='support source', ax=axes[1]);
sns.scatterplot(data=df, x='mean feature importance', y='PValue', hue='support source',
edgecolor=None, alpha=0.5, markers=['.', '.', 'X'], style='support source', ax=axes[2]);
sns.pairplot(df, hue="support source", markers=['.', '.', 'X'],
vars=["mean feature importance", "logFC", "mean nFDR"],
plot_kws=dict(edgecolor=None, alpha=0.25));
/home/tyler/anaconda3/envs/gsfenv/lib/python3.7/site-packages/seaborn/distributions.py:305: UserWarning: Dataset has 0 variance; skipping density estimate.
warnings.warn(msg, UserWarning)
UMAP Embeddings of Selections¶
counts, labels = gsf.get_gem_data(union_coll, selected_gene_sets=['Boruta_treatment'], count_variable='qt_counts',
annotation_variables=['treatment', 'genotype', 'time'])
mapper = umap.UMAP(densmap=True, random_state=50, metric='manhattan').fit(counts.values)
fig, axes = plt.subplots(1, 3, figsize=(21, 7))
umap.plot.points(mapper, labels=labels['treatment'], background='black', ax=axes[0], color_key_cmap='Set1');
umap.plot.points(mapper, labels=labels['genotype'], background='black', ax=axes[1], color_key_cmap='Set2');
umap.plot.points(mapper, labels=labels['time'], background='black', ax=axes[2], color_key_cmap='plasma');
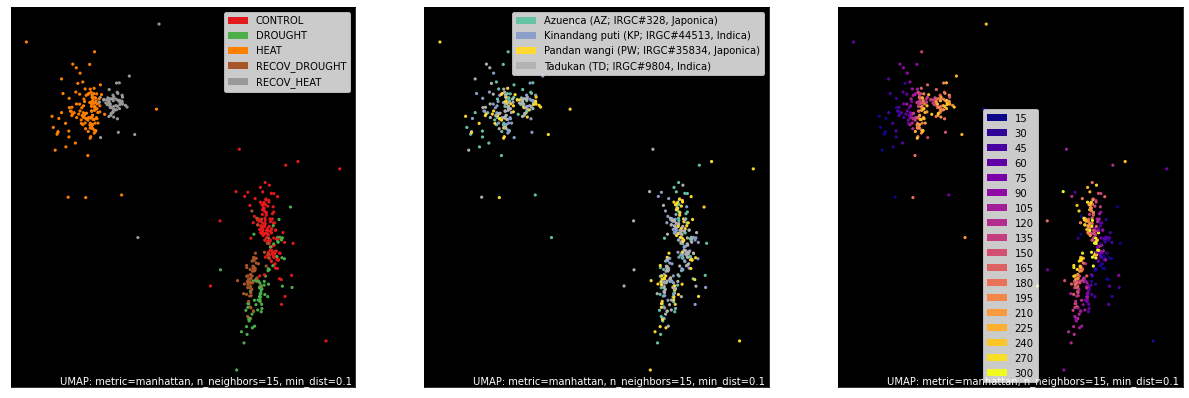
counts, labels = gsf.get_gem_data(union_coll, selected_gene_sets=['literature_union'], count_variable='qt_counts',
annotation_variables=['treatment', 'genotype', 'time'])
mapper = umap.UMAP(densmap=True, random_state=50, metric='manhattan').fit(counts.values)
fig, axes = plt.subplots(1, 3, figsize=(21, 7))
umap.plot.points(mapper, labels=labels['treatment'], background='black', ax=axes[0], color_key_cmap='Set1');
umap.plot.points(mapper, labels=labels['genotype'], background='black', ax=axes[1], color_key_cmap='Set2');
umap.plot.points(mapper, labels=labels['time'], background='black', ax=axes[2], color_key_cmap='plasma');
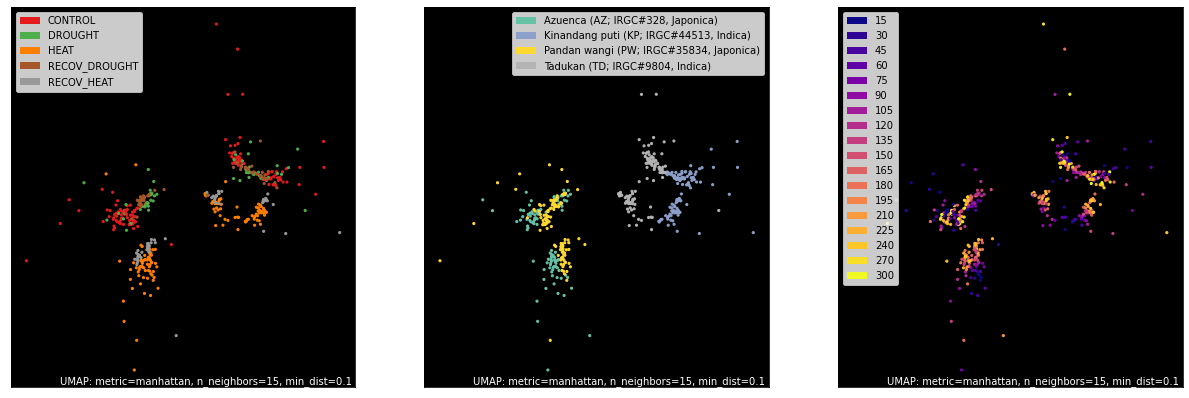
Clustermap Selection¶
def series_to_colors(series, cmap, categorical=True):
keys = series.unique()
colors = hv.plotting.util.process_cmap(cmap, len(keys), categorical=categorical)
mapper = {k: c for k, c in zip(keys, colors)}
return series.map(mapper)
# az_samples = agem.data.sel(Sample=agem.data.genotype == 'Azuenca (AZ; IRGC#328, Japonica)')['Sample'].values
df, labels = gsf.get_gem_data(union_coll,
# sample_subset=az_samples,
annotation_variables=['treatment', 'genotype'],
selected_gene_sets=["Boruta_treatment",
"literature_union"],
gene_set_mode='intersection',
count_transform=lambda counts: np.log2(counts.where(counts > 0)),
output_type="pandas")
color_df = pd.DataFrame({
"treatment": series_to_colors(labels['treatment'], "Set1"),
"genotype": series_to_colors(labels['genotype'], "Set2")
})
sns.clustermap(df.fillna(0),
metric='cityblock',
row_colors=color_df,
row_cluster=False,
dendrogram_ratio=0.1,
cmap='jet',
figsize=(10, 10));
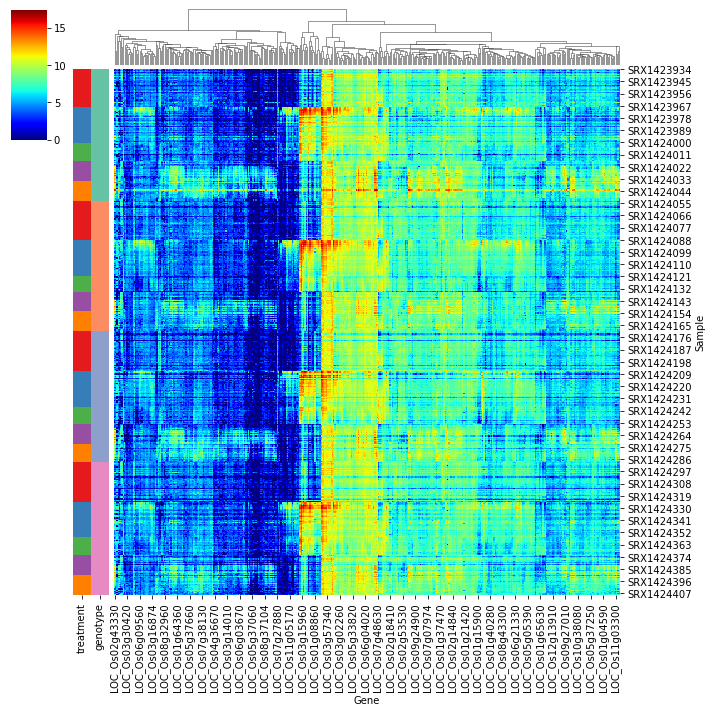
Functional Enrichment / Gene Set Enrichment Analysis¶
In investigating a set of genes it is common to examine available annotations for those genes. With special interest given to those annotations that appear enriched within our selection.
GSEAPY is useful here.
import gseapy as gp
from gseapy.plot import gseaplot
func_data_path = OSF_PATH.joinpath('GEMmakerGEMs', 'raw_annotation_data', 'oryza_sativa.MSU_v7_0.genes2annots.txt')
fdf = pd.read_csv(func_data_path, sep='\t', low_memory=False)
fdf = fdf[fdf['Gene'].isin(agem.data.Gene.values)]
fdf.head()
| Gene | Term | Definition | CV | Reaction-id | EC | Protein-id | Protein-name | Gene-id | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | LOC_Os12g04990 | PWY-321 | cutin biosynthesis | PlantCyc | RXN-16389 | EC-6.2.1.3 | GN7F-26073-MONOMER | NaN | GN7F-26073 |
| 1 | LOC_Os11g35400 | PWY-321 | cutin biosynthesis | PlantCyc | RXN-16389 | EC-6.2.1.3 | GN7F-17597-MONOMER | NaN | GN7F-17597 |
| 2 | LOC_Os05g04170 | PWY-321 | cutin biosynthesis | PlantCyc | RXN-16389 | EC-6.2.1.3 | GN7F-17804-MONOMER | NaN | GN7F-17804 |
| 3 | LOC_Os06g06350 | PWY-321 | cutin biosynthesis | PlantCyc | RXN-16389 | EC-6.2.1.3 | GN7F-18230-MONOMER | NaN | GN7F-18230 |
| 4 | LOC_Os05g25310 | PWY-321 | cutin biosynthesis | PlantCyc | RXN-16389 | EC-6.2.1.3 | GN7F-26550-MONOMER | NaN | GN7F-26550 |
support = dge_collection["'0 + treatment'__treatment[HEAT]"].get_support()
term_counts = fdf.loc[fdf['Gene'].isin(support)].groupby('Term')['Gene'].count()
term_counts = term_counts[term_counts > 10]
scores = dge_collection["'0 + treatment'__treatment[HEAT]"].data['logFC'].sel(Gene=support).to_dataframe()
term_genes = dict()
for term in term_counts.index.values:
term_genes[term] = fdf.groupby('Term')['Gene'].unique()[term]
pre_res = gp.prerank(rnk=scores,
gene_sets=term_genes,
processes=4,
permutation_num=100, # reduce number to speed up testing
outdir='prerank',
format='png', seed=6)
pre_res.res2d.sort_index().head()
| es | nes | pval | fdr | geneset_size | matched_size | genes | ledge_genes | |
|---|---|---|---|---|---|---|---|---|
| Term | ||||||||
| GO:0005524 | 0.287134 | 0.933461 | 0.543210 | 0.540046 | 2218 | 15 | LOC_Os04g01740;LOC_Os05g44340;LOC_Os06g12370;L... | LOC_Os04g01740;LOC_Os05g44340;LOC_Os06g12370 |
| IPR002068 | 0.821481 | 2.909723 | 0.000000 | 0.000000 | 39 | 17 | LOC_Os03g14180;LOC_Os11g13980;LOC_Os01g04370;L... | LOC_Os03g14180;LOC_Os11g13980;LOC_Os01g04370;L... |
| IPR008978 | 0.793451 | 2.646324 | 0.000000 | 0.000000 | 50 | 20 | LOC_Os03g14180;LOC_Os11g13980;LOC_Os01g04370;L... | LOC_Os03g14180;LOC_Os11g13980;LOC_Os01g04370;L... |
| expressed protein | 0.355513 | 1.429853 | 0.106383 | 0.117277 | 14309 | 38 | LOC_Os07g47840;LOC_Os02g15930;LOC_Os11g05170;L... | LOC_Os07g47840;LOC_Os02g15930;LOC_Os11g05170;L... |
| path:dosa04141 | 0.670797 | 2.644200 | 0.000000 | 0.000000 | 199 | 27 | LOC_Os03g14180;LOC_Os11g13980;LOC_Os01g04370;L... | LOC_Os03g14180;LOC_Os11g13980;LOC_Os01g04370;L... |
terms = pre_res.res2d.index
terms
Index(['IPR002068', 'IPR008978', 'path:dosa04141', 'expressed protein',
'GO:0005524'],
dtype='object', name='Term')
gseaplot(rank_metric=pre_res.ranking, term=terms[0], **pre_res.results[terms[0]])
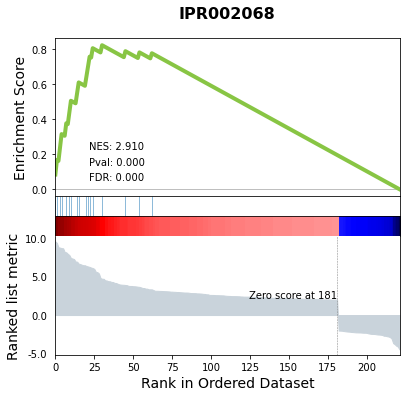
Conclusion & Next Steps¶
Having introduced the GSForge package by example we hope you are motivated to compare and share gene selection methodologies. Recall that the scope of gene / feature selection is unique to each experiment, no gold standard method or statistical measure yet exists to unambiguously interpret RNA-seq data. In most cases researchers or their collaborators are interested in a reduced list of genes (rather than the entire genome!) from which to form a hypothesis.
We hope you feel free to contribute, share and question methodologies, and that you find GSForge helpful in this endevour.
References¶
Wilkins, O. et al. EGRINs (Environmental gene regulatory influence networks) in rice that function in the response to water deficit, high temperature, and agricultural environments. Plant Cell 28, 2365–2384 (2016).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. (2018).
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R. & Pfister, H. UpSet: Visualization of Intersecting Sets Europe PMC Funders Group. IEEE Trans Vis Comput Graph 20, 1983–1992 (2014).