Viewing Gene Expression Distributions¶
A great deal of quality-control information comes from the alignment step itself. Here we are concerned with how the data looks as an ensemble. Many methods are particularly concerned with the dispersion of genes.
In this notebook we demonstrate the plotting utilities provided by gsforge to examine such distributions.
Set up the notebook
import itertools
import holoviews as hv
import numpy as np
import xarray as xr
import GSForge as gsf
hv.extension('bokeh')
import matplotlib.pyplot as plt
import colorcet as cc
from datashader.mpl_ext import dsshow, alpha_colormap
import datashader
from functools import partial
import seaborn as sns
from mpl_toolkits.axes_grid1 import make_axes_locatable


Declare used paths
# OS-independent path management.
from os import environ
from pathlib import Path
OSF_PATH = Path(environ.get("GSFORGE_DEMO_DATA", default="~/GSForge_demo_data/")).expanduser().joinpath("osfstorage", "oryza_sativa")
NORMED_GEM_PATH = OSF_PATH.joinpath("AnnotatedGEMs", "oryza_sativa_hisat2_normed.nc")
Load an AnnotatedGEM
agem = gsf.AnnotatedGEM(NORMED_GEM_PATH)
agem
<GSForge.AnnotatedGEM>
Name: Oryza sativa
Selected GEM Variable: 'counts'
Gene 55986
Sample 475
View available count arrays
agem.count_array_names
['counts',
'TPM_counts',
'uq_raw_counts',
'uq_tpm_counts',
'quantile_counts',
'tmm_counts']
gsf.plots.gem.GenewiseAggregateScatter(
agem,
count_variable='counts',
x_axis_selector='mean',
y_axis_selector='variance',
count_transform=lambda c: np.log2(c + 1),
datashade=True,
dynspread=True,
)
Recall that all gsforge plotting operations allow use of the Interface data selection pipeline.
In this case we can select another count array and view the normalized distributions.
gsf.plots.gem.GenewiseAggregateScatter(
agem,
count_variable='TPM_counts',
x_axis_selector='mean',
y_axis_selector='variance',
count_transform=lambda c: np.log2(c + 1),
datashade=True,
dynspread=True,
)
# for count_var, y_axis in itertools.product(agem.count_array_names, ['variance', 'fano', 'cv_squared']):
# plot = gsf.plots.gem.GenewiseAggregateScatter(
# agem,
# count_variable=count_var,
# x_axis_selector='mean',
# y_axis_selector=y_axis,
# axis_transform=('log 2', lambda ds: np.log2(ds.where(ds > 0))),
# datashade=True,
# dynspread=True,
# )
# hv.save(plot, f'figures/genewise_aggs/gw_agg_{count_var}_log2_mean_vs_log2_{y_axis}.png', dpi=300, toolbar=None)
For some reason the adjoint png files produced have extra white space. We can remove that with a solution from github
# from PIL import Image
# from PIL import ImageOps
# padding = 5
# padding = np.asarray([-1*padding, -1*padding, padding, padding])
# for figure in Path('figures/genewise_aggs').glob('gw_agg_*.png'):
# image = Image.open(figure)
# image.load()
# imageSize = image.size
# # remove alpha channel
# invert_im = image.convert("RGB")
# # invert image (so that white is 0)
# invert_im = ImageOps.invert(invert_im)
# imageBox = invert_im.getbbox()
# imageBox = tuple(np.asarray(imageBox)+padding)
# cropped = image.crop(imageBox)
# cropped.save(figure)
Grouped-Sample Covariance¶
These plotting functions can take a few minutes to complete.
treatment_labels = agem.data['treatment'].to_series().unique()
treatment_labels
array(['CONTROL', 'HEAT', 'RECOV_HEAT', 'DROUGHT', 'RECOV_DROUGHT'],
dtype=object)
# %%time
# for group_a, group_b in itertools.combinations(treatment_labels, 2):
# plot = gsf.plots.gem.GroupedGeneCovariance(agem, group_variable='treatment',
# x_group_label=group_a, y_group_label=group_b,
# count_transform=lambda c: np.log(c + 0.25)
# ).opts(size=0.75, width=300, height=300)
# hv.save(plot, f'figures/grouped_covariance/covariance_{group_a}_vs_{group_b}.png', 'png')
Sample-wise Distributions¶
These plotting func tions can take a few minutes to complete.
Kernel Density Estimates¶
# %%time
# for count_var, hue in itertools.product(['counts'], [None]):
# plot = gsf.plots.gem.SamplewiseDistributions(agem, count_variable=count_var, hue_key=hue,
# datashade=False)#.opts(width=300, height=300)
# hv.save(plot, f'figures/kde/samplewise_kde_{count_var}_{hue}.png', toolbar=None)
Empirical Cumulative Distribution¶
# %%time
# for count_var, hue in itertools.product(agem.count_array_names[:-1], [None, 'treatment', 'genotype']):
# plot = gsf.plots.gem.EmpiricalCumulativeDistribution(agem, hue_key=hue, count_variable=count_var, datashade=True)
# hv.save(plot, f'figures/ecdf/ECDF_{count_var}_{hue}.png', dpi=300, toolbar=None)
plt.style.use('default')
plt.rcParams.update({'font.size': 11, 'font.family': 'serif'})
log_counts, _ = gsf.get_gem_data(agem, count_transform=lambda c: np.log2(c + 1), count_variable='TPM_counts')
data = xr.Dataset({
"mean": np.mean(log_counts, axis=0),
"variance": np.sqrt(np.var(log_counts, axis=0)),
})
data = data.set_coords(['mean', 'variance'])
hv.operation.datashader.datashade(
hv.Points(data, kdims=['mean', 'variance']),
cmap='inferno', cnorm='log',
).opts(bgcolor='lightgrey', padding=0.05, width=600, height=400, show_grid=True)
log_counts, _ = gsf.get_gem_data(agem, count_transform=lambda c: np.log2(c + 1))
data = xr.Dataset({
"mean": np.mean(log_counts, axis=0),
"variance": np.sqrt(np.var(log_counts, axis=0)),
})
data = data.set_coords(['mean', 'variance'])
hv.operation.datashader.datashade(
hv.Points(data, kdims=['mean', 'variance']),
cmap='inferno', cnorm='log',
).opts(bgcolor='lightgrey', padding=0.05, width=600, height=400, show_grid=True)
log_counts, _ = gsf.get_gem_data(agem, count_transform=lambda c: np.log2(c + 1), count_variable='quantile_counts')
data = xr.Dataset({
"mean": np.mean(log_counts, axis=0),
"variance": np.sqrt(np.var(log_counts, axis=0)),
})
data = data.set_coords(['mean', 'variance'])
hv.operation.datashader.datashade(
hv.Points(data, kdims=['mean', 'variance']),
cmap='inferno', cnorm='log',
).opts(bgcolor='lightgrey', padding=0.05, width=600, height=400, show_grid=True)
/home/tyler/anaconda3/envs/gsfenv/lib/python3.7/site-packages/xarray/core/computation.py:742: RuntimeWarning: invalid value encountered in log2
result_data = func(*input_data)
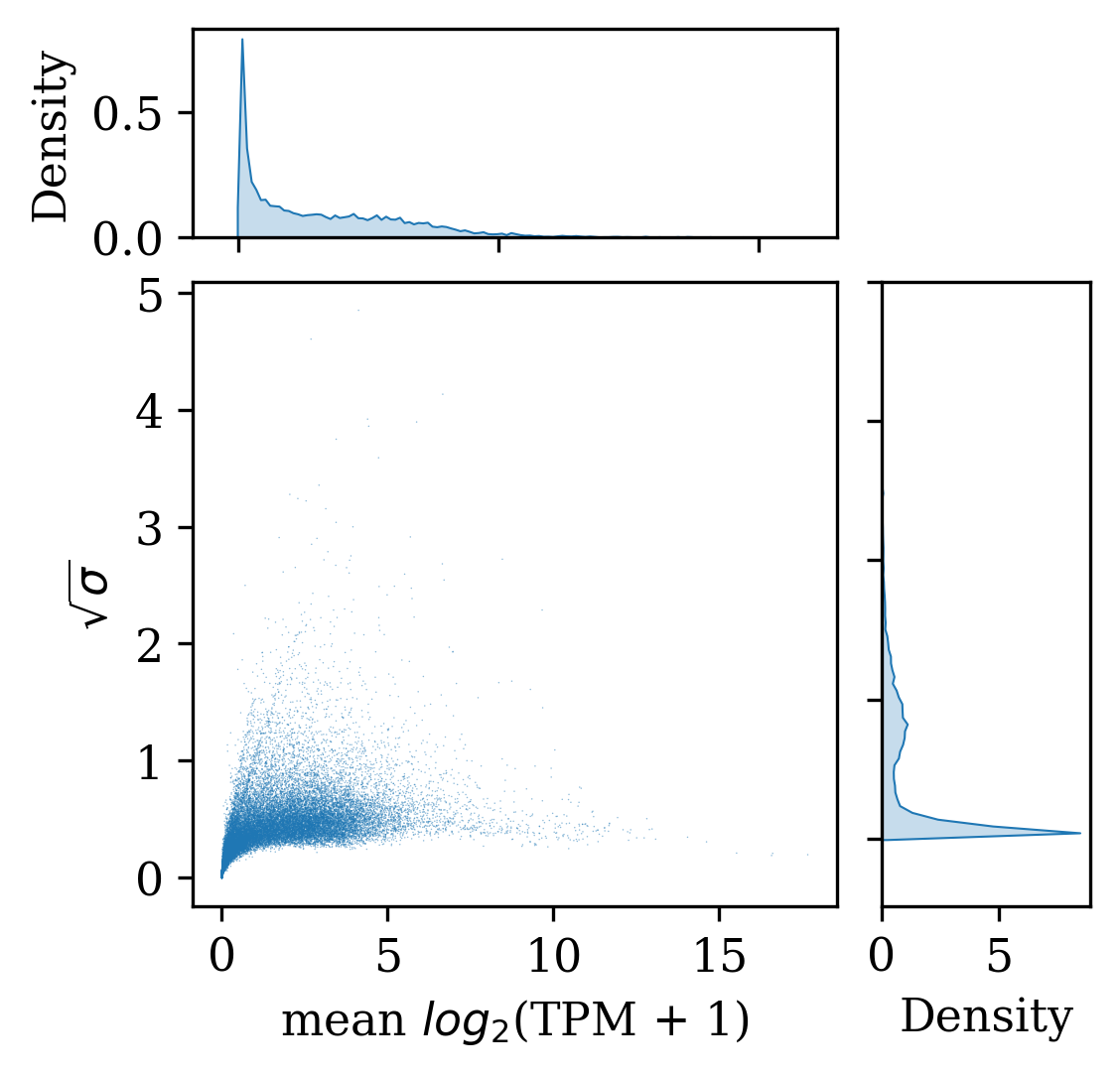
log_counts, _ = gsf.get_gem_data(agem, count_transform=lambda c: np.log2(c + 1), count_variable='TPM_counts')
data = xr.Dataset({
"mean log2 counts": np.mean(log_counts, axis=0),
"sqrt log2 variance": np.sqrt(np.var(log_counts, axis=0)),
})
data = data.set_coords(['mean log2 counts', 'sqrt log2 variance'])
x_min = data['mean log2 counts'].values.min()
x_max = data['mean log2 counts'].values.max()
y_min = data['sqrt log2 variance'].values.min()
y_max = data['sqrt log2 variance'].values.max()
padding = 0.05
x_pad = (x_max - x_min) * padding
y_pad = (y_max - y_min) * padding
x_min -= x_pad
# x_max += x_pad
x_max = 11.5
y_min -= y_pad
# y_max += y_pad
y_max = 2
fig_inches = 3.5
fig_dpi = 300
fig_pixels = int(fig_inches * fig_dpi)
fig, axScatter = plt.subplots(figsize=(fig_inches, fig_inches), constrained_layout=True, dpi=fig_dpi)
axScatter.scatter(
data['mean log2 counts'].values,
data['sqrt log2 variance'].values,
s=0.5,
alpha=0.5,
marker='.',
edgecolors='none',
)
# dsshow(
# data,
# datashader.Point('mean log2 counts', 'sqrt log2 variance'),
# shade_hook=datashader.transfer_functions.dynspread,
# norm='eq_hist', cmap="Blues", ax=axScatter,
# aspect='auto',
# x_range=(x_min, x_max),
# y_range=(y_min, y_max),
# plot_width=fig_pixels,
# plot_height=fig_pixels,
# );
divider = make_axes_locatable(axScatter)
axHistx = divider.append_axes("top", 0.2*fig_inches, pad=0.15)
axHisty = divider.append_axes("right", 0.2*fig_inches, pad=0.15)
axHistx.xaxis.set_tick_params(labelbottom=False)
axHisty.yaxis.set_tick_params(labelleft=False)
axHistx.set_xlim(x_min, x_max)
axHisty.set_ylim(y_min, y_max)
sns.kdeplot(data["mean log2 counts"].values, ax=axHistx, linewidth=0.5, shade=True, bw_adjust=0.05)
sns.kdeplot(y=data["sqrt log2 variance"].values, ax=axHisty, linewidth=0.5, shade=True, bw_adjust=0.05)
axScatter.set(xlabel='mean $log_2$(TPM + 1)', ylabel='$\sqrt{\sigma}$');
[Text(0.5, 0, 'mean $log_2$(TPM + 1)'), Text(0, 0.5, '$\\sqrt{\\sigma}$')]